
Diario da Universidade de Vigo
Froito dun traballo multidisciplinar desenvolvido por investigadores das universidades de Vigo e San
Paso adiante na implementación de algoritmos máis eficientes no aliñamento de secuencias xenéticas
Permite avanzar na resolución dun problema computacional con implicacións matemáticas e bioinformáti

Juan José Nieto, Ángela Torres e Iván Area

Teorema principal
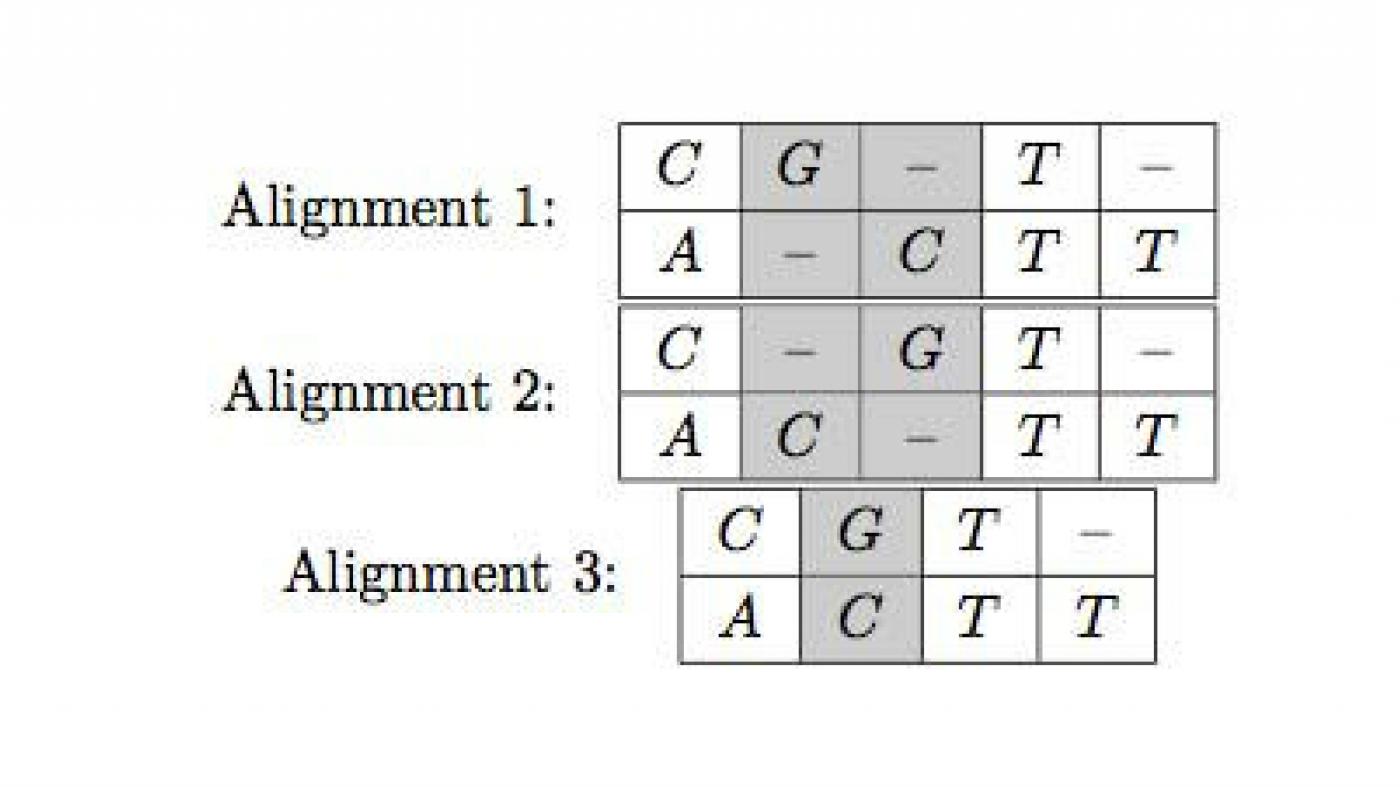
Tipos de aliñamento
A revista BMC Bioinformatics vén de publicar o traballo multidisciplinar desenvolvido polos investigadores da Universidade de Vigo Iván Area e da Universidade de Santiago de Compostela Helena Andrade, Juan José Nieto Roig e Ángela Torres, titulado The number of reduced alignments between two DNA sequences, que permite avanzar no coñecemento das secuencias xenéticas e que pode dar lugar á implementación de algoritmos máis eficientes, a través de fórmulas explícitas e un reducido custo computacional. O estudo que ten a súa orixe na tese de doutoramento de Helena Andrade, é froito da colaboración de investigadores e investigadoras, que levan moitos anos traballando en distintos campos e que acordaron unir esforzos para resolver un problema concreto, que está relacionado coa comparación de dúas secuencias xenéticas. “Sabíase que existen moitos xeitos de aliñar dúas secuencias, tantos que non era posible enumerar todos desde o punto de vista computacional. Agora sabemos exactamente cantas hai, sexa cal sexa a súa lonxitude, que na práctica pode ser moi grande”, explica Iván Area, profesor do Departamento de Matemática Aplicada II da Universidade de Vigo.
Este punto de encontro entre Bioloxía e Computación que é a bioinformática, ten repercusións directas na mellora da calidade da vida humana a través, por exemplo, da detección e tratamento de enfermidades e a produción de alimentos xeneticamente modificados, “usando ferramentas de sistemas de computación, ou a colección, organización, almacenamento e recuperación da información biolóxica que se atopa en bases de datos”, detalla Area. No caso concreto da comparación de secuencias de ADN (ácido desoxirribonucleico) no que se centra o traballo publicado en BMC Bioinformatics, esta ten moitas aplicacións, como por exemplo, dar información sobre a funcionalidade dun xene. “O virus H5N1 é unha variante do virus da gripe aviaria, que os expertos consideran que pode mudar nunha forma transmisible entre humanos. Ten moléculas de ARN, ácido ribonucleico, e dúas delas son HA e PB1, a primeira delas é especialmente importante na transmisibilidade e a PB1 na súa virulencia. Xa que logo, saber se as mutacións manteñen estas moléculas é altamente importante”, explica a profesora Ángela Torres da Facultade de Medicina da Universidade de Santiago de Compostela.
Secuencias de ADN e cadeas matemáticas
O ADN, que contén instrucións xenéticas usadas no desenvolvemento dos organismos vivos e é responsable da transmisión hereditaria, está formado por dous longos polímeros de unidades sinxelas, chamadas nucleótidos: adedina, citosina, guanina ou timina. Para comparar dúas secuencias, o punto de partida é considerar cada unha delas como unha cadea ou secuencia matemática onde cada unha das entradas pode ser calquera dos catro nucleótidos. “Hai distintos xeitos de comparar dúas secuencias xenéticas. Dado que algúns aliñamentos son moi similares, podemos considerar equivalentes algúns deles e iso reduce o número e o custo computacional. A pregunta á canto o reduce, e a fórmula obtida neste traballo permite calcular exactamente esa diferenza”, explica Iván Area.Aínda que os investigadores das universidades de Vigo e Santiago recoñecen que neste ámbito de traballo aínda quedan outros problemas por resolver, lembran que na actualidade existen distintos algoritmos implementados para obter o aliñamento óptimo, entre dúas secuencias, por exemplo no European Bioinformatics Institute. “Non obstante o noso traballo pode dar lugar á implementación de algoritmos máis eficientes xa que damos fórmulas explícitas e con pouco custo computacional”, conclúe o catedrático Juan José Nieto Roig.